저번 글에 RNA-seq 데이터 전처리와 관련해 포스팅을 하였다.
이번편에서는 실제 논문을 따라한 예시 실습을 올리고자 한다.
※단 주의할 점은 이번편에 사용한 데이터는 #microarry data이다.
그치만, DEG 분석과정은 RNA-seq과 동일하게 시행하였다 (DEG 분석에 더 focusing하길)
이번편은 한마디로,
#GEOquery tool을 사용해 GEO dataset을 뽑아내고 #DESeq2 tool로 DEG분석하는 법이다.
내가 simulation한 논문의 제목은 'Identification of DEGs-related prognostic risk model for survival prediction in breast carcinoma patients'이다.
논문 링크 ↓

Since the imbalance of gene expression has been demonstrated to tightly related to breast cancer (BRCA) genesis and growth, common genes expressed of BRCA were screened to explore the essence in-between. In current work, most common differentially expressed genes (DEGs) in various subtypes of BRCA w...
pubmed.ncbi.nlm.nih.gov
논문 분석 내용은 다음과 같다.
5개의 GSE dataset(Spain, Taiwan, Singapore, USA, mexico)에 대해 DEG 분석을 통해 Tumor/Normal sample 간의 upregulated/down regulated 차이가 나는 gene을 finding하고 survival 분석까지 진행하였다.
분석(3):
1. DEG Analysis
2. Enrichment analysis (biological pathway 확인)
3. Kaplan-Meir 분석을 통한 생존 상관성 확인
이번 편에서는 #DEG 분석에 대한 #R 프로그래밍 실습 코드만 넣어보고자 한다.
실습을 하기 이전에 데이터를 어디에서 불러오냐?가 궁금할 것이다.
[1] Input data
이번 실습에 사용한 database는 #GEO database이다.
그러나 GEO에서 절대 수동으로 다운로드 받을 필요가 없다!
이번엔 GEOquery라는 library를 사용해서 GEO data를 불러올 것이니 밑에는 참고만 하자
GEO database에 대해 부가 설명을 하자면
GEO database
NCBI에서 주관해서 만들어진 database이다. 대용량의 RNA-seq, Microarry, NGS data등을 제공해주는 곳으로 GEO ID별로 저장된 database를 찾을 수 있다.
출처 입력
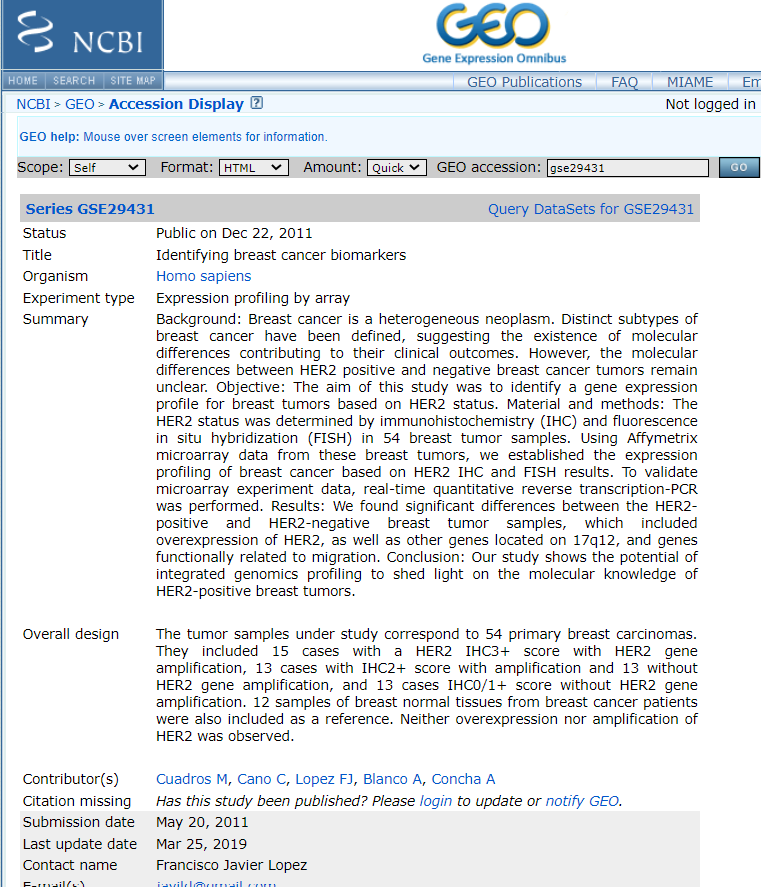
위와 같이 GEO accession number를 입력하면
해당 데이터의 publication된 논문의 내용과 실험에 대한 자세한 내용이 적혀있다.
여기서 찾아 보면 좋은 내용은
1.data 종류이다. RNA-seq 인지, microarray인지 epigenomics인지 확인
2, 실험 기법, 왜냐면 나중에 reference 달 때이 affy로 했는지 등에 따라 name labeling이 달라지기 때문이다.
그 외에도 필요한 부가적인 내용을 꼼꼼히 보면 된다.
[2] R code - data calling, DEG alanysis, Visualization
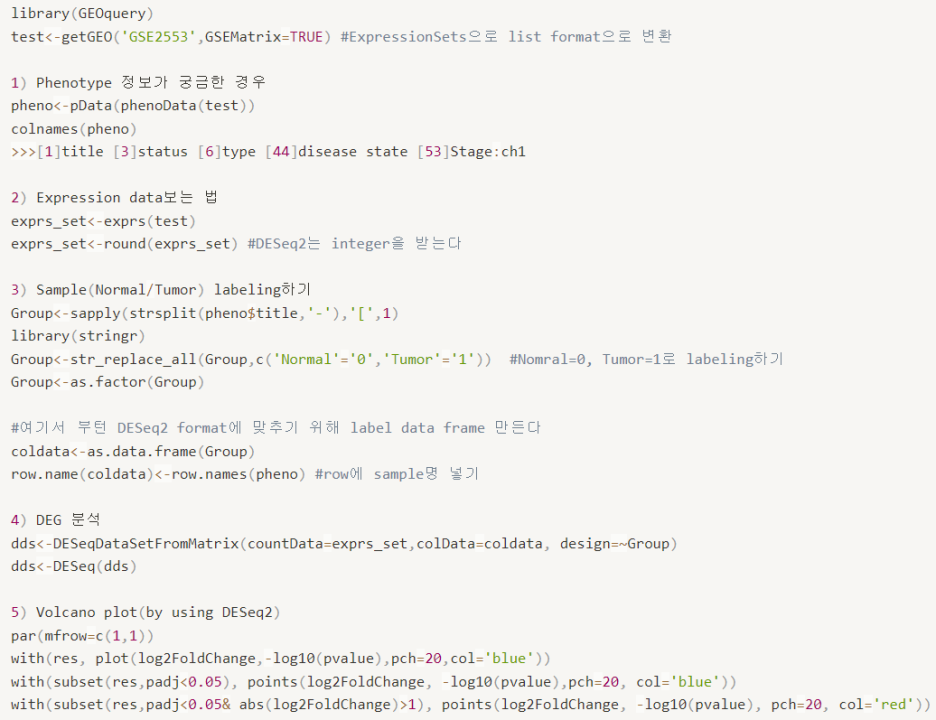
R code
위 논문의 GEO accession (GEO2553)에 대한
DEG 분석과 visualization 코드이다.
위에서 'getGEO' 명령어를 통해 database를 불러왔다면
어떤 데이터가 있는지 궁금할 것이다.
각 데이터 베이스마다 format은 다르겠지만
여기서 확인할 중요한 것은 두가지이다.
1) expr(RNA-seq) => 발현 데이터
2) pData(phenoData(RNA-seq) => 표현형 데이터
이다.
분석 이전에 RNA-seq 데이터의 구성에 대해 얘기하자면
아래와 같다.
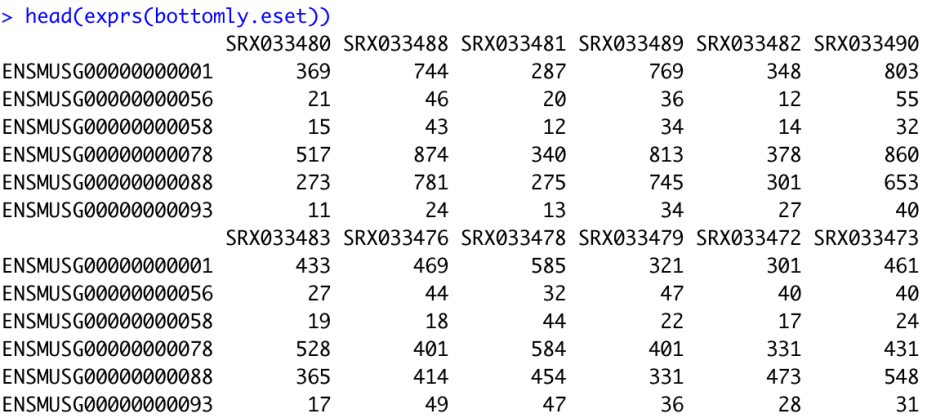
RNA-seq expression data
column은 sample을 얘기하고,
row는 gene이다 (ENSM? 하여튼 뭔 형식으로 변경한 것이다)
DESeq2 library를 통해 분석하기 이전에
phenotype을 0/1로 binary 형식으로 바꿔줘야 하기에 labeling 변경해야 한다.

visualization
시각화 결과이다.
윗줄은 논문의 volcano plot 결과,
밑에는 내가 그린 volcano plot을 비교한 것이다.
전처리가 조금 다른지 확별히 같지는 않다
데이터 해석은 크게 2가지를 알면 된다.
X축-Log2FoldChange 는 Case와 control 군의 차이 나는 유전자를 나타내고
Y축- (-LogPvalue)는 유전자 내에서 P-value값이 크게 나는 유전자를 나타낸다.
case/control이란 암환자와 정상인군의 차이나는 유전자를 말한다.
따라서 X축 오른쪽 Y축 위를 의미하는 것은 up-regulated gene,
왼쪽, 아래의미는 down-regulated gene들을 의미한다.