목차:
[1] Heatmap, Hirarchical Clustering
[2] K-means Clustering
[3] Normalization & Batch Effect Removal
[1] Heatmap
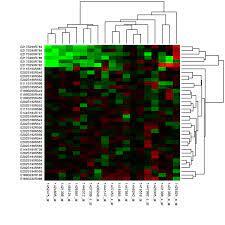
유전자를 clustering한다는 것 = 다른 sample들 사이에 비슷한 expression profile을 가짐을 의미함
Sample들을 clustering한다는 것 = clustering을한 유전자들이 비슷한 expression profile을 가짐을 의미
- Clustering을 하는 이유?
첫번 째, data 특징 혹은 실험 오류를 파악하는데 도움을 줌 → differential expression
두번 째, sample들을 classify하는 데 사용 가능
Cluster quality 평가하는 법: cluster끼리의 distance를 측정 ( 가까울 수록 비슷함 )
Cluster stability 평가하는 법 : 몇몇 데이터를 지워봐도 cluster가 유지되는지 혹은 noise 값을 추가했을 때, 몇몇개의 parameter값이 변경 혹은 유지되는지 확인
Hierarchical Clustering

과정 - gene 1이 gene 2 ~ 8까지의 distance를 계산함, gene 2도 나머지 유전자들과 거리 계산 해 가장 적은 distance를 가지는 유전자들끼리 연결해준다 (node 연결)
[2] K-means Algorithm

- 방법:
1) 임의적으로 K centroids(별 모양)을 선택함
2) 데이터를 군집에 할당하는 것으로 위에서 정한 중심점을 중심으로 데이터를 그 중심값의 군집으로 할당한다.
3) 중심(centroid) 재조정하는 단계로 위에서 만든 군집중에 가장 중간에 위치한 지점으로 중심점을 재설정한다.
4) cluster가 변하지 않을 때까지 1~3번을 반복한다.
- 특징:
1) Determisitc한 method이다.
-> 첫 번재 cluster center값이 매우 중요함, 잘못 지정된 경우 local optimal 값에 trap될 수 있기 때문
2) initial cluster center값을 어떻게 선정?
-> hierarchical clustering를 running해서 cut line을 선정하거나 다른 initial center값으로 여러번 시행한다.
- 고려해야될 점:
K값 선정!을 고려해야함 ( K는 군집 개수를 의미함 )
좋은 cluster란, cluster값은 작으면서 군집간의 길이는 서로 먼 것이다.
K값을 선정하는 equation도 있음 (참고)

[3] Normalization & Batch effect
- Normalization과 Batch Effect의 차이점
Normalization은 library 제작과정에서 생긴 차이, gene length 차이, 대규모의 dropout을 제거 혹은 대체하기 위한 방법
Batch effect는 실험 설계, 실험 수행(sequencing 기계 차이, 실험 시간 차이, 실험실 차이) 등에서 오는 차이를 줄이기 위해 하는 처리
- Normalization 방법
- Median Scaling
- LOESS (LOcally WEighted Scatterplot Smoothing, local linear fits)
- Quantile Normalization
→ 많이 쓰이는 정규화 방법 중 하나임.
→ 방법: 각 quantile의 평균값 측정 → sample값을 quantil mean값으로 대체

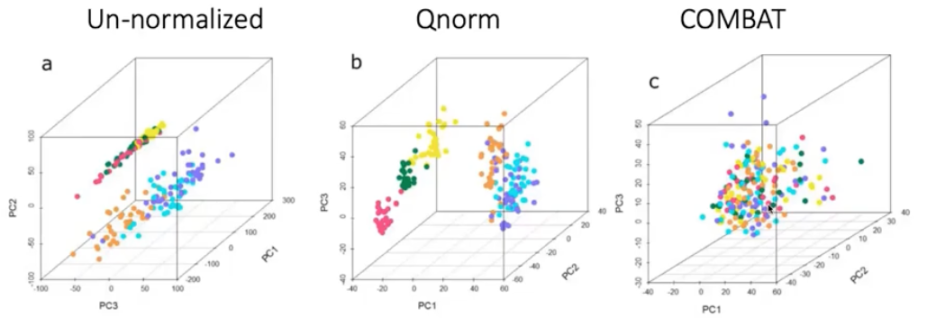
- Batch effect란?
: 서로 다른 연구나 실험 혹은 실험 과정에서 온 세포들 혹은 샘플들을 합칠 때 생기는 기술적 차이를 제거한는 것임
→ 방법으로는 Quantile normalization 기법, PCA 기법(tool: COMBAT), limma tool을 활용해서도 제거할 수 있다.
→ 얻는 효과? batch effect를 제거하고 나서 DEG분석을 하면 더 명확한 유전적 차이를 발견할 수 있다.