※ Index
1. scRNA-seq 기본용어
2. scRNA 실험 techinques
3. scRNA 전체 분석 과정
4. 분석 방법: data preprocess(QC) → normalization → batch effect
나는 공부할 때 scRNA-seq이 RNA-seq보다 특별한 점이 뭐길래 쓰지? 란 관심을 가졌고,
scRNA 데이터를 이해하기 위해선 scRNA 실험의 특징에 대해 이해할 필요를 느끼게 되었다.
실험을 통해 나온 scRNA-seq 데이터의 특징을 작성하고,
본격적으로 데이터를 분석하는 방법에 대해 공유하고 한다.
#scRNA 기본 용어
scRNA 데이터를 알기 위해선 기본적으로 알아야 할 용어들이 있다.
바로 UMIs (Unique Molecular Identifiers)와 barcode란 용어들로,
실험상에서 의미하는바를 알아야 데이터에서 뭘 의미하는지 알 수 있다.
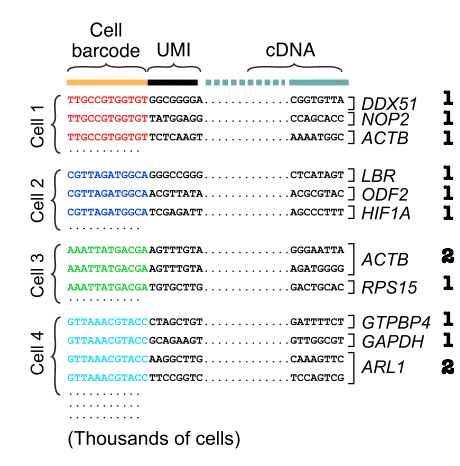
간단히 정립하면 UMI = mRNA이고, barcode = cell type이다.
조금 더 detail적으로 UMI의 의미와 용도는
짧은 DNA molecules에 dna fragment의 끝에 붙이는 tag로. 용도는 PCR로 나온 중복들을 제거하기 위해 붙이기 위함이다.
#scRNA technique
scRNA를 분석하기 전 대략적으로 scRNA의 실험 기법엔 무엇이 있는지 알아야 나중에 분석할 때 도움이 된다.
scRNA techinque들로는 10X Chromium, SMART-seq2, drop-seq, seq-well uses microwells 등이 있고, 10X Chromium이 가장 많이 쓰인다.
실험 방식의 차이는 cell을 isolate하는 방식으로, 크게 droplet(물과 기름으로 분리) 하는 것과 plate-based으로 분리하는 방법이 있다.
대표적인 #10X Chromium (2017)의 실험 과정에 대해서 기입하도록 하겠다.

Droplet encapsulation 방식으로 세포를 기름방울(oil droplet)에 가둬서 단일세포로 분리하는 방법이다.
가장 왼쪽에 10x Barcoded Gel beads 안에가 DNA 끝에 위에서 설명한 barcode와 UMI가 부착된 DNA molecules들이 들어있는 것이고, 하나의 molecule이 지나가면서 cell이 만나서 방울을 만드는 방식이다.
#scRNA 전체 분석 과정

RNA의 전체 분석 과정 workflow는 다음과 같다.
왼쪽을 보면 preprocessing이 적혀있고, 오른쪽에 downstream analysis가 적혀있다.
왼쪽 part에서 실험을 통해 scRNA의 각 cell별 count matrices가 나오면
normalization과 quality control를 진행해야 한다.
Normalization과 QC를 진행하는 이유는 실험에서 섞이는 오류를 제거하기 위함이고,
더 나아가 Feature selection을 통해 주요한 gene들만 뽑아내는 과정을 거치면 본격적으로 분석할 수 있는 단계이다.
추후 분석 과정에 대해선 다음 2편에 작성하도록 하겠다.
#전처리: QC / Normalization / Batch effect
실험을 통해 나온 데이터들을 보면
0인 value값들이 많이 나온 것을 확인 할 수 있다.
0인 value들은 missing data를 뜻하는 것으로 특정 gene에서는 모든 cell type들이 0이 나오는 것들도 있다.
이러한 0처리는 제외시켜야 다음 분석 단계에서 조금 더 accurate한 결과들을 얻을 수 있다.
#Quality Control

quality control를 하는 방법은
위 그림처럼 count per barcodes depth에 대한 frequency를 본다.
0~4,000 사이의 depth를 살펴보면
1,600 count전까지는 매우 낮은 frequency를 볼 수 있다.
(이 낮은 frequency들을 제외시키는 이유는 20 cells보다 적게
발현되는 유전자들의 경우 추후 cluster 분석에서
cluster 되기 어렵기 때문에 제외시키는 것이다)
이 1,600 counts 전 것은 outlier로 선정해 모두 제외시킨다.
outlier는 dead cells, broken membrances, doublets들에 해당한다.
#Normalization
normalization할 때 쓰는 library로는 SCnorm이 있고 regression 방법을 통해
표준화시키는 것이다.
bulk RNA seq으로 부터 얻어진 scRNA-seq의 경우 normalization은
TPM을 많이 쓰고
일반적인 분석의 경우엔 RPKM을 많이 사용한다.
또한 normalization하고 나서log(x+1)로 transformed를 시행하는 데
이를 시행하는 이유는
1) log transformed expression value들은 log fold change를 의미하고
2) 데이터의 비대칭도를 줄여 다음 단계 분석에서 normally 분포할 수 있도록 도와주기 위해서이다
#Batch correction
batch correction을 할 수 있는 tool로 가장 많이 알려진 것은
Combat으로 linear regression 기반으로 batch correction을 해준다.
batch correction을 해주는 이유는 transcriptome상의 cell cycle의 영향을 제거하기 위함이다.
