이번엔 준비된 scRNA는 어떤 과정으로 분석하는지 작성해보고자 한다.
scRNA computational analysis 과정은 다음과 같다.
normalization → batch effect removal → dimension reduction → clustering → cell annotation이다
이번 편은 Dimension reduction을 왜 해야되는지,
dimension reduction하는 방법 3가지에 대해 공유하고자 한다.
#Dimension reduction
을 하는 이유는 25,000여개에 달하는 유전자들 중에서 중요한 유전자("HVG": highly variable genes)들을 뽑아내기 위해서이다. 많은 유전자들 중에선 zero-count인 유전자들이 많고 이 모든 유전자들을 다 쓸 필요가 없기 때문에 feature selection을 통해 제거해야 한다.
방법으로는 크게 3가지가 있다.
linear한 방법인 PCA(Principal Component Analysis), non-linear한 방식 tSNE 그리고 graph방식도 있다.
1) PCA: PCA is a linear approach that generates reduced dimensions by maximizing the captured residual variance in each further dimension
2) tSNE: focus on capturing local similarity at the expense of global structure
( non-linear projection 방법 → visualization에 유용)
3) UMAP: tSNE와 비슷한 방법이지만 better preserves global structure (전체적인 구조를 보기에 더 유용)
- PCA와 t-SNE의 특징과 단점:
- PCA의 문제점은 선형 분석 방식으로 값을 사영하기 때문에 차원이 감소되면서, 군집화 되어 있는 데이터들이 뭉게져 제대로 구별할 수 없다는 문제를 가진다.
- t-SNE의 원리는 선택한 점과 다른 점까지의 거리를 측정해서 친밀도값을 측정해 서로 가까운 값으로 묶는 것인데, 단점으로는 매번 계산할 때마다 축의 위치가 바뀌어 다른 모양으로 나타나는 머신러닝 모델의 feature extraction으로 사용하기엔 어려운 점이 있다.
#Clustering
- Clustering 방법:
1) K-means (가장 기본, 오래된 방법)
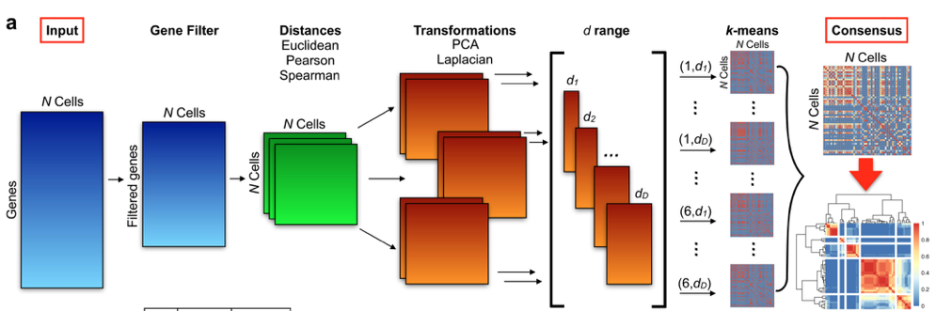
2) single-cell consensus clustering (SC3) → 여러개로 k-menas를 시행하고 합침, 단점으로는 큰 데이터를 사용하기엔 어렵고 시간이 오래걸림
3) hierarchical clustering
4) graph clustering
나는 공부하면서 scRNA-seq을 clustering을 왜 해야 하는지에 대해 궁금증이 생겼다.
- scRNA-seq을 clustering을 하는 이유?
→ clustering을 잘하면 sparse한 data도 이해하기 쉬워진다. cell annotation을 하기 위한 것이 가장 큰 목적인데, 해당 유전자가 cell annotate 되면 어떤 역할을 하는지 알 수 있다.
clustering을 하고 나면 cell annotation을 한다.
여기서도 생겼던 궁금점은 이미 cell 단위로 isolate한 것인데 왜 또 마지막에 gene에 cell을 define할까?였다.
이에 대해 찾아 보니
→ scRNA input data는 여러가지의 cell들의 transcriptomes 정보를 가지고 있는건 맞지만 그들의 identity가 뭔지는 모르는 상태이다.
→ single cell은 단일 세포 수준에서 유전자 발현을 측정하는 만큼, 정확하게 정량하기 어렵다. 특히 발현량이 낮은 유전자들은 적은 수의 세포를 증폭하기 때문에 아예 count가 안되는 유전자들이 많다('Dropout')
→single cell 한개만 보고 분석하게 되면 false가 많아 clustering을 통해 해당 cell의 identity를 annotate해주는 것이 더 정확하다.